மரபுக் கடத்தல் கொள்கைகள் மற்றும் மாறுபாடுகள் - மென்டலின் குறைபாடுகள் | 12th Zoology : Chapter 4 : Principles of Inheritance and Variation
12 ஆம் வகுப்பு விலங்கியல் : அத்தியாயம் 4 : மரபுக் கடத்தல் கொள்கைகள் மற்றும் மாறுபாடுகள்
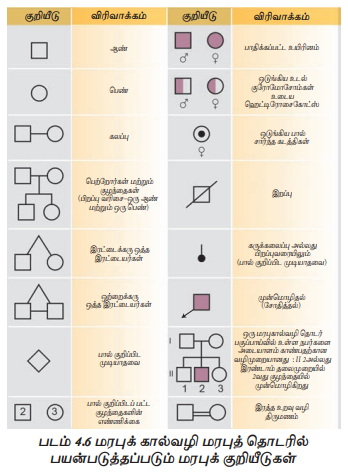
மென்டலின் குறைபாடுகள்
மென்டலின் குறைபாடுகள் (Mendalian disorders)
ஒரு மரபணுவில் ஏற்படுகின்ற மறுசீரமைப்பு அல்லது திடீர்மாற்றம், மெண்டலின் குறைபாட்டை ஏற்படுத்துகின்றன. மெண்டலின் மரபுக்கடத்தல் விதிகளின் படியே இவை சேய் உயிரிகளுக்குக் கடத்தப்படுகின்றன. தலாசீமியா, அல்பினிசம், பினைல் கீட்டோநீயூரியா, அரிவாள் செல் இரத்த சோகை நோய் மற்றும் ஹன்டிங்டன் கோரியா போன்றவை மென்டலியன் குறைபாடுகளுக்கு எடுத்து காட்டுகளாகும் இந்த வகையான குறைபாடுகள், ஓங்கு தன்மை அல்லது ஒடுங்குதன்மை கொண்டோ மற்றும் உடல் குரோமோசோம் அல்லது பால் குரோமோசோம் சார்ந்த பண்பாகவோ இருக்கலாம்.
தலாசீமியா (Thalassemia)
இது உடல் குரோமோசோமில் உள்ள ஒரு ஒடுங்கு பண்பு மரபணுவின் திடீர் மாற்றத்தினால் ஏற்படும் நோயாகும். இந்நோயினால், இரத்த சிவப்பணுக்கள் அதிகமாக சிதைக்கப்படுகின்றன. இயல்புக்கு மாறான ஹீமோகுளோபின் மூலக்கூறுகள் உருவாவதே இதற்குக் காரணமாகும். இயல்பான ஹீமோகுளோபின் நான்கு பாலிப்பெப்டைடு சங்கிலியால் ஆனது அதில் 2 ஆல்பா மற்றும் 2 பீட்டா குளோபின் சங்கிலிகளாகும். தலசீமியா நோயால் பாதிக்கப்பட்டவர்களின் ஆல்பா அல்லது பீட்டா சங்கிலிகளில் ஏதாவதென்று பாதிக்கப்பட்டுள்ளதால் இயல்புக்கு மாறான ஹீமோகுளோபின் மூலக்கூறுகள் உருவாகி, இரத்த சோகையை ஏற்படுத்துகிறது.
பாதிக்கப்பட்டுள்ள ஹீமோகுளோபின் சங்கிலி வகையின் அடிப்படையில் ஆல்பா மற்றும் பீட்டா தலசீமியா என இரு வகைகளாகப் பிரிக்கலாம். 16ஆம் குரோமோசோமில் நெருக்கமாக அமைந்த HBA1 மற்றும் HBA2 ஆகிய இரண்டு ஜீன்கள் தலாசீமியாவை கட்டுப்படுத்துகின்றன. திடீர்மாற்றம் அல்லது நீக்கம் அடைந்த ஒன்று அல்லது ஒன்றுக்குமேற்பட்ட ஆல்பா மரபணுக்கள் ஆல்ஃபா தலாசீமீயாவை உண்டாக்குகின்றன. பீட்டா தலசீமியா என்பது பீட்டா குளோபின் சங்கிலி உற்பத்தி பாதிப்படைவதால் ஏற்படுகிறது. இதனை குரோமோசோம் பல் உள்ள ஒற்றை ஜீன் (HBB) கட்டுப்படுத்துகிறது. பொதுவாக காணப்படும் இவ்வகை தலாசீமியா கூலியின் இரத்தசோகை (Cooley's anaemia) எனவும் அழைக்கப்படுகிறது. இந்நோயினால் ஆல்பா சங்கிலி உற்பத்தி அதிகரித்து இரத்த சிவப்பணுக்களின் சவ்வுகள் சேதமுறுகின்றன.
பினைல்கீடோநியூரியா
இது பினைல் அலனைன் வளர்சிதை மாற்ற பிறவிக் குறைபாட்டு நோயாகும் (Inborn error of metabolism). உடல் குரோமோ சோம்களில் உள்ள ஒரு இணை ஒடுங்கு மரபணுக்களால் இந்நோய் ஏற்படுகிறது. குரோமோசோம் 12ல் அமைந்துள்ள பினைல் அலனைன் ஹைட்ராக் ஸிலேஸ் என்ற கல்லீரல் நொதியை சுரப்பதற்குக் காரணமான PAH மரபணுவின் திடீர்மாற்றத்தால் இந்நோய் உண்டாகிறது. பினைல் அலனைனை டைரோசினாக மாற்ற இந்நொதி அவசியமாகும். இந்நோயால் பாதிக்கப்பட்டவர்களுக்கு இந்நொதி சுரக்காது. இதனால் தேங்கிய பினைல் அலனைன்கள் பினைல் பைருவிக் அமிலமாகவும் மற்றும் அதன் வழிப்பொருளாகவும் மாறுகின்றன. இதன் விளைவால் அதிதீவிர மூளை குறைபாட்டு நோய், தோல் மற்றும் முடிகளில் குறைவான நிறமிகள் ஆகியவை உண்டாகின்றன. பினைல் பைருவிக் அமிலம் சிறுநீர் வழியாக வெளியேற்றப்படுகிறது.

நிறமி குறைபாட்டு நோய் (Albinism)
நிறமிகுறைபாட்டு நோய் ஒரு வளர்சிதை மாற்ற பிறவி குறைபாட்டு நோயாகும் (Inborn error of metabolism). இவை உடற்குரோமோசோமில் உள்ள ஒடுங்கிய ஜீனால் ஏற்படுகிறது. தோலின் நிறத்திற்கு மெலானின் நிறமிகள் காரணமாக உள்ளன. மெலானின் நிறமி இல்லாத நிலை 'நிறமி குறைபாட்டு நோய்' என அழைக்கப்படுகின்றது. ஒரு நபர், ஒடுங்கிய அல்லீல்களை பெற்றிருக்கும்போது, டைரோசினேஸ் நொதியை உற்பத்தி செய்ய முடியாது. மெலானோசைட் செல்களில் உள்ள டைஹைட்ராக்ஸி பினைல் அலனைனை (DOPA) மெலானின் நிறமியாக மாற்ற இந்நொதி தேவைப்படுகின்றது. இந்நோயால் பாதிக்கப்பட்ட நபர்களின் தோல், உரோமம், ஐரிஸ் மற்றும் பல பகுதிகளில் இயல்பான எண்ணிக்கையில் மெலானோசைட் செல்கள் காணப்படும். ஆனால் அவற்றில் மெலானின் நிறமி இருப்பதில்லை.
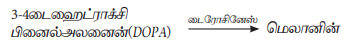
ஹன்டிங்டன் கோரியா
இது மனிதனில் உடற்குரோமோசோமின் ஓங்கு தன்மை கொண்ட கொல்லி மரபணுவால் ஏற்படுகிறது. தன்னியல்பான உடல் நடுக்கம் மற்றும் படிப்படியான நரம்பு மண்டல சிதைவு , அதனுடன் மனநிலை பாதிப்பு மற்றும் உடல்பலம் குன்றல் ஆகியன இந்நோயின் பண்புகளாகும். இந்நோய் கொண்ட நபர்கள் 35 முதல் 40 வயதுக்கிடையே இறப்பை சந்திக்கிறார்கள்.